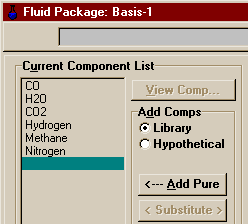
This example will take you through the entire process of setting up multiple reactions and creating a plug flow reactor in HYSYS (as shown in the picture above). The same example problem has been worked in Aspen and Matlab. A completed case has been pre-built and is located in the file PlugFlowEx.hsc in the \\Hartsook\Hysys\SAMP403 directory, though I recommend you work through the example yourself so that you do not miss anything. The reactions that we are going to model are those encountered in the early stages of the production of such chemicals as ammonia and methanol, both of which may start with a Natural Gas feed. Here are the two reactions that we shall consider: CO + H2O <--> H2 + CO2 The first reaction is the reforming reaction in which the natural gas (here modeled entirely as methane) is reacted with steam to form hydrogen and carbon monoxide. The second reaction is the Water Gas Shift Reaction in which yet more hydrogen is produced and the CO changed to CO2 (this is especially important in ammonia sysnthesis as both CO and CO2 will poison the catalyst used to make ammonia from hydrogen and nitrogen, and the CO is harder to remove than CO2). Note that the Water Gas Shift is an equilibrium reaction and is modeled as a reversible reaction. The reactor that we will model is like one of the Water Gas Shift Reactors in the ammonia process (occurs after the reforming reactors), and at the considerably lower temperature at which we shall be operating, we should expect to see mostly the second reaction and little of the first. In fact, we won't see any, but if we were modeling this whole process, we would need to account for the possible effects of both reactions in the shift reactors as well as the reforming reactors. Here, the purpose is to demonstrate another kinetic form in HYSYS. To start, open a new case in HYSYS and add a new fluid package. Choose Antoine for the Property Package. The example calls for an ideal system (we'd like to be able to compare to Matlab and Aspen which we cannot do if differing thermodynamics are used). Unfortunately, HYSYS does not have a package labeled ideal. However, according to Chapter 8 of RV1 (link only good from AL-C126), the vapour pressure models (such as Antoine) are for essentially ideal systems. Alternatively, you might try comparing those results to the results of an activity coefficient model like Margules with its vapour model set to ideal and all of its binary coefficients (both Aij's and Bij's) set to zero. Switch to the Components Page and install the following components into the fliud package:
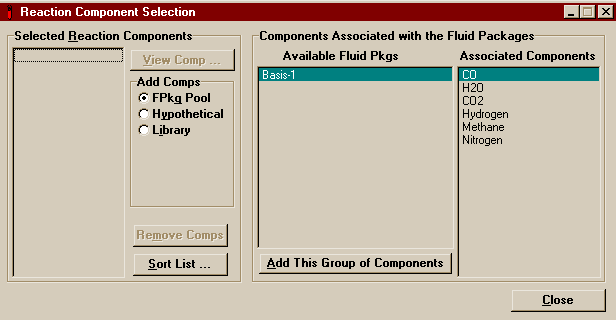
Return to the Simulation Basis Manager window and switch to the Reactions page. The first thing that must be done is tell HYSYS which components are to be available for reaction, so push the button marked Add Comps... . A window should open that looks like this.
Press Add This Group of Components and then close the window. Note that if you had not already created the fluid package, you could simply have switched to the Library radio button in the add components group and added the components as you had added them to the fluid package. In fact, when the reaction is later associated with a specific fluid package, if the reaction components were not already listed among the package's components, they would be added then. Now on the Reactions page of the Simulation Basis Manager, press the Add Rxn... button.
r2 = DBED*A*e(-Ea2/(R*T))*(yc*yw - yd*yh/Keq2)
As mentioned above, the first reaction is Langmuir-Hinshelwood, so highlight that in the Reactions window and press Add Reaction. Fill out the form that appears with components and coefficients so that it looks like the picture below then press the Basis tab to move to that page.
Note that unlike the second
reaction (which has an equal number of moles on both sides
and therefore shouldn't), the first reaction contains
pressure terms. You will note, however, that in each
instance it is a singular mole fraction multiplied by the
overall pressure. This is equivalent to the partial pressure
of the species. Therefore you should change your basis to
Partial Pres. You may leave the base component as
Methane (note the base component of a reaction must always
be a reactant for that reaction, though it may be the
product of a different reaction).
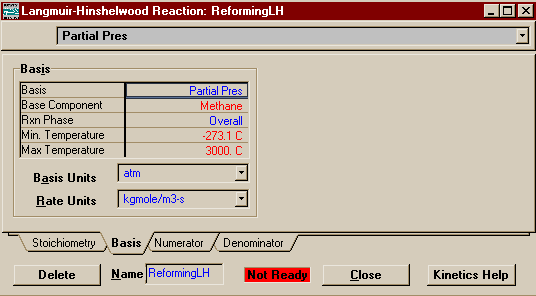
Since we anticipate that these reactions will only take
place at temperatures where the entire process stream is
gaseous, a reaction phase of either Vapour Phase or
Overall is fine. When you are all finished the Basis Page should look like this:
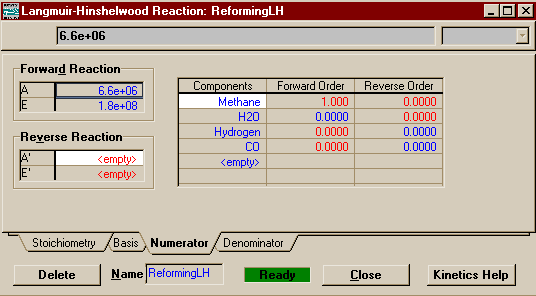
On the Numerator Page, you shall begin filling in the information that will tell HYSYS the actual form of the rate equation. If you have not already done so, I suggest that you push Kinetics Help, which will call up the following window:
The various basis functions in the reaction I have colored yellow and the parts that become the A's are in red. You can see that in this numerator there is no reverse
reaction and that the forward reaction is first order in
methane and zero order in steam (the latter is a trifle
suspect and indicates that some assumptions were probably
made about the ratio of steam to carbon and that the steam
partial pressure was probably incorporated into the rate
constant). Therefore, in the components matrix, you should
change it so that all orders for both forward and reverse
reactions are zero, except for the forward order of Methane
which should remain 1.
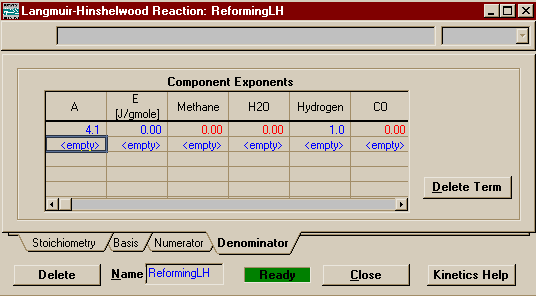
Note that the status has changed to Ready. Do not believe it! Though HYSYS does not require it, we still need to input the Denominator (and it would be kind of silly to use the Langmuir-Hinshelwood form if you didn't have a Denominator). Moving to the denominator page, the form of input changes slightly. It is now in matrix form, because you have the ability to put in as many terms as you want into the denominator, each with differing A's, E's, and bases to go with them. Enter Kh = 4.053 into A, set E equal to 0, and put a 1 under Hydrogen.
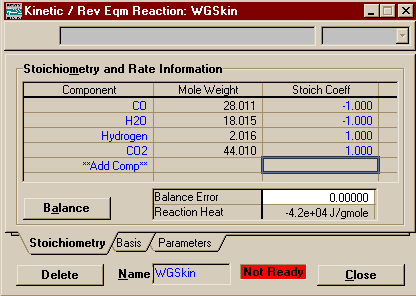
You are done entering the data for the first reaction. You may close this window. Go back to the little Reactions window and add a Kinetics (Rev Eqm) reaction. If you closed the Reactions window you may get it back by going to the Simulation Basis Manager and pushing Add Rxn on the Reactions Page. Add the components and the stoiciometric coefficients like we did for the first reaction, then move to the Basis Page.
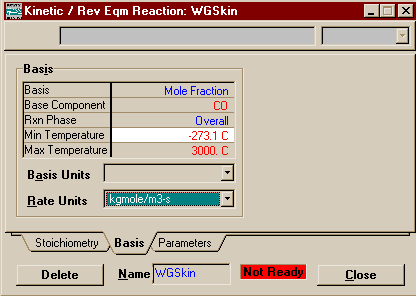
On the Basis Page, set the basis to Mole Fraction, the reaction phase to Overall or Vapour Phase, and the reaction units to kgmole/m3-s.
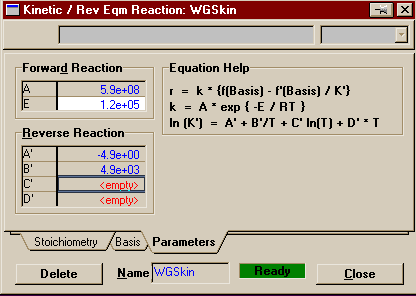
Moving on to the Parameters Page, we see that this time the Kinetics Help is already on the page. Below I have copied the second reaction rate equation. This time, as with the first reaction, the pre-exponential factor is in red and the bases are in yellow. Note that this time, however, you are not asked for
information on the orders of the components in the forward
and backward reaction. That is because this formulation of
rate equation can only be used with elementary reactions
(remember those are reactions where the orders are equal to
the coefficients). The reason being that that is how
Keq's are defined.
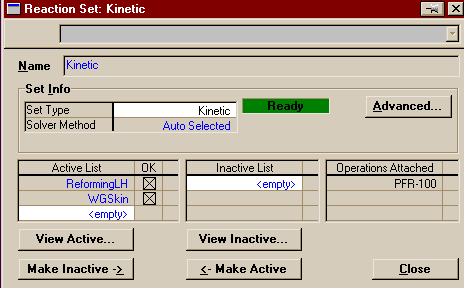
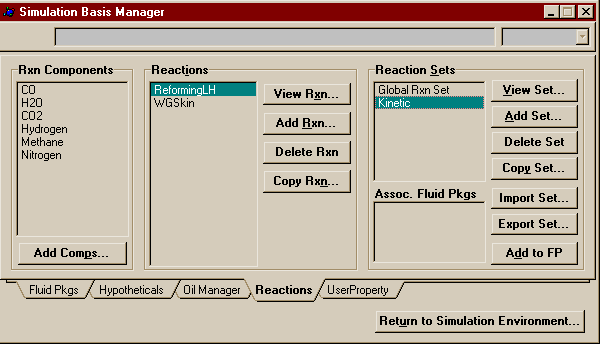
You are now finished entering the second and final reaction. You may close this window and return to the Reactions Page of the Simulation Basis Manager. Now that you are done creating your reactions, you must add them to a reaction set. Reaction Sets are groups of one or more reactions. They may be exported and imported. They must be attached to fluid packages to be used in the flowsheets to which those fluid packages are associated. You can attach as many as you like to a fluid package, and thus use different sets on different units within the flowsheets. Reaction Sets may be used with reactors, columns, and separators. Conversion reactions cannot be placed in the same set as other types of reaction. For more information on Reaction Sets see Section 11.4 in RV1 (link only good from AL-C126). To start, press the Add Set button in the Reaction Sets Group, add both of your newly created reactions to the Active List. Note that the Global Rxn Set already contains the two reactions created, but practice is good for you and this way you get to see the Reaction Set window.
Close the above window and look back at the Simulation Basis Manager, which should now look like this:
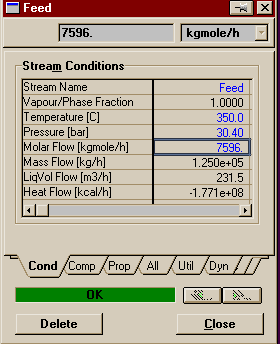
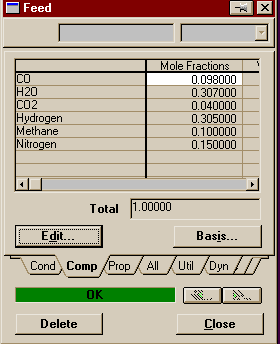
Make sure your reaction set is highlighted and press Add to FP. A window will pop up listing all the available fluid packages (in this case, there should be only one). Select it and press Add Set to Fluid Package. Your fluid package should now appear under Assoc. Fluid Pkgs when your reaction set is selected. You have now completed everything you need to do in the basis environment. You may now enter the Simulation Enironment. Begin by creating two material streams on the PFD. Name one Feed and the other Product. On Feed's property view, set the temperature to 350o C, the pressure to 30 atm, and the total flowrate to 2110 moles/s (7596 kgmoles/hr). Fill out the compositions as shown below.
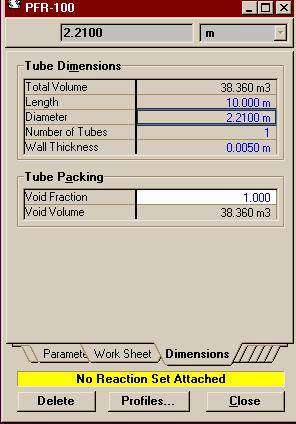
Open the property view of the reactor. If you have already connected the streams to the reactor then the Connections Page will be completed (note that unless you connect a heat stream, the reactor is assumed to be adiabatic by HYSYS). You may move to the Parameters Page. The only thing you need to do there is set the Pressure Drop to 0. Move two tabs over to the Dimensions Page. Fill in the settings as they are below.
These are the same dimensions as those used in the
Aspen example (with
the same correction in the diameter for porosity that you
can read about there).
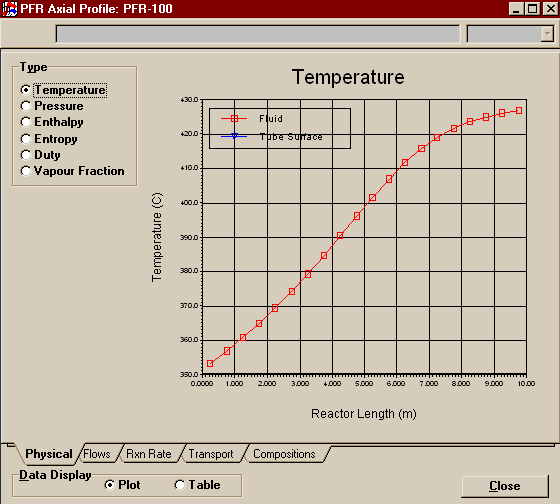
To see some of the results of your reactor, click on the Profiles button at the bottom of its Property View.
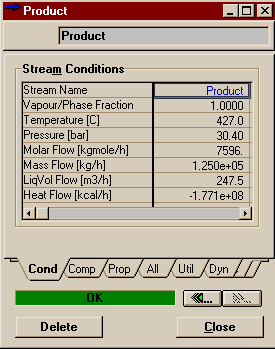
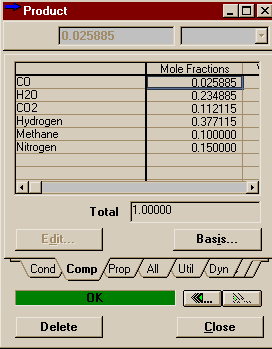
Above is one of the reactor profiles you can choose. It is important that you note -- it's easier to see in table mode than graph mode -- that the data points at the ends say they are for .25 m and .95 m, not 0 m and 10 m like you might expect. You have to remember that HYSYS integrates over the length of the reactor by dividing it into 20 sub-volumes (that was the default on the Reactions Page of the Property View and we left it at that). Ten meters divided into twenty groups means .5 m per division. The point that HYSYS records is thus halfway between the divisions, but is valid in HYSYS for the whole sub-volume. Thus the temperature, composition, etc. of the product stream contains the same values as 9.75 m down the reactor. In other words, don't worry that HYSYS is leaving off its charts the first and last values. The first point corresponds to the reactor inlet and the last point to the reactor outlet. Below are the results you should have gotten for your Product stream.
Compare these to the results we found in the Aspen simulation. Agreement is good in both the mole fractions and the final temperature (off by about a degree). These results correspond to a thermodynamic package of Antoine. If you were to go back and change this to Margules, set the vapour model to ideal, and delete all the binary parameters, you would find that the results hardly change at all (the final temperature changes by 0.2 degrees Celcius). Thus, it is likely that the difference between the HYSYS and Aspen models are the result of column integration differences versus property package differences. |
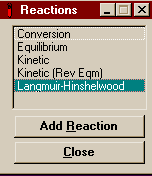 This is the window that comes
up. There are five forms of reaction that you can enter in
HYSYS. To know which ones to use you must know the form of
the kinetic data you have. In this example we will be
looking at the last two forms of reaction. For a description
of all of the different reaction types, see
Chapter 11 of RV1
(link only good from AL-C126). Reaction 1 is the reforming
reaction and is in the form that may be modeled easily by
Langmuir-Hinshelwood. The Langmuir-Hinshelwood
equation you may (or may not) remember from CENG 390 is the
form used for heterogeneous catalysis when you must worry
about products or inerts using up active sites on the solid
catalyst. Reaction 2, the Water Gas Shift reaction, uses the
standard Kinetic (Rev Eqm) form, in which you know
the forward reaction constant and use the equilibrium
constant to get the reverse reaction constant. Below is all
the information we are given on the nature of the
reactions.
This is the window that comes
up. There are five forms of reaction that you can enter in
HYSYS. To know which ones to use you must know the form of
the kinetic data you have. In this example we will be
looking at the last two forms of reaction. For a description
of all of the different reaction types, see
Chapter 11 of RV1
(link only good from AL-C126). Reaction 1 is the reforming
reaction and is in the form that may be modeled easily by
Langmuir-Hinshelwood. The Langmuir-Hinshelwood
equation you may (or may not) remember from CENG 390 is the
form used for heterogeneous catalysis when you must worry
about products or inerts using up active sites on the solid
catalyst. Reaction 2, the Water Gas Shift reaction, uses the
standard Kinetic (Rev Eqm) form, in which you know
the forward reaction constant and use the equilibrium
constant to get the reverse reaction constant. Below is all
the information we are given on the nature of the
reactions.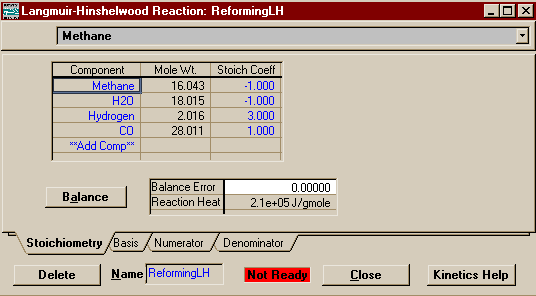

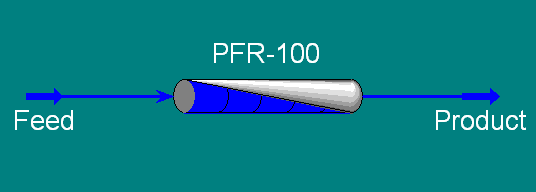
 Now,
from either the Oblect Palette or the Unit Ops Page of the
Workbook, add a Plug Flow Reactor to the flowsheet and
connect the Feed and Product streams (remember a shortcut
for toggling into and out of the connecting mode is to hold
down <Ctrl>). Your PFD should look like the one
below.
Now,
from either the Oblect Palette or the Unit Ops Page of the
Workbook, add a Plug Flow Reactor to the flowsheet and
connect the Feed and Product streams (remember a shortcut
for toggling into and out of the connecting mode is to hold
down <Ctrl>). Your PFD should look like the one
below.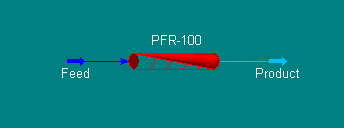
Return to the Reactions Page | Return to the HYSYS Page | Return to the CENG 403 homepage