 Home
Home![]() Agarose can become superheated and violently boil over. Exercise caution when heating. Swirl flask occasionally during heating.
Heat until close inspection reveals that the agarose is 100% dissolved. Undissolved agarose will appear as little flecks that look like Lilliputian contact lenses.
Agarose can become superheated and violently boil over. Exercise caution when heating. Swirl flask occasionally during heating.
Heat until close inspection reveals that the agarose is 100% dissolved. Undissolved agarose will appear as little flecks that look like Lilliputian contact lenses.
![]() Ethidium bromide is a powerful mutagen and is moderately toxic and should be handled with care.
Ethidium bromide is a powerful mutagen and is moderately toxic and should be handled with care.
Note: Concentrated ethidium bromide solutions should be decontaminated. One method is to treat 0.5 µg/ml staining solutions of EtBr with 1 g/liter activated charcoal, filter and incinerate the residue. Slurries of activated charcoal can be used to decontaminate surfaces (see Maniatis et al, (1989) for additional methods of decontamination).
|
1. |
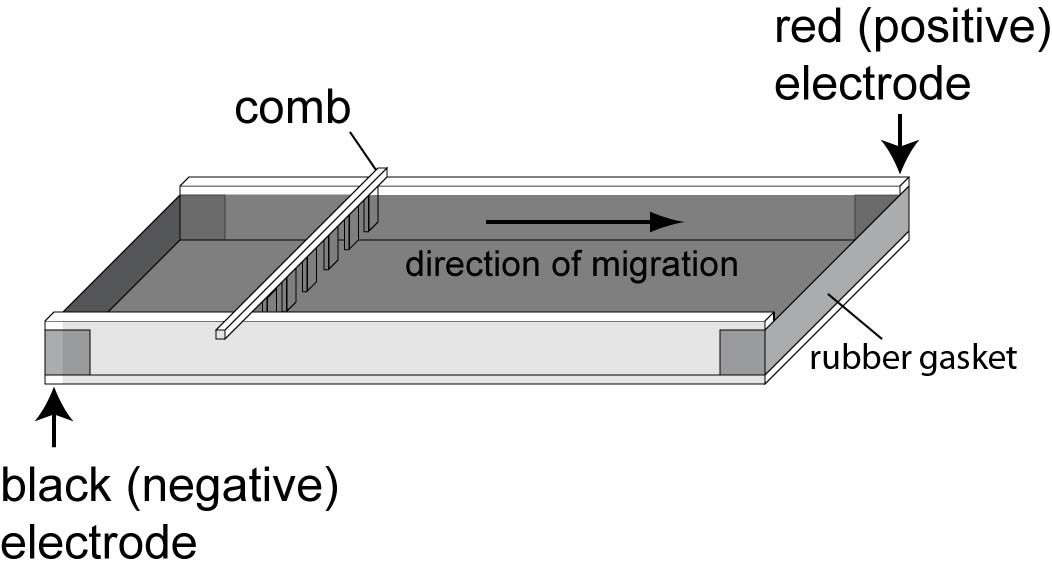
Prepare gel tray as diagrammed using an adjustable gel caster. Level the tray using a bubble level. |
|
|
|
2. |
Flasks of completely melted 1% agarose in 1X TBE have been prepared and the melted solution is incubating at 50°C. Carefully pour melted agarose (60 ml) into a beaker. Remember the solution is hot!! |
|
|
|
3. |
After the instructor adds EtBr to the gel, gently swirl the agarose. (Let the instructor know when you are ready to add the EtBr to your gel.) |
|
|
|
4. |
Immediately, pour the melted agarose into the level casting tray. Use a pipet tip to push bubbles towards the bottom of the gel. |
|
|
|
5. |
Let the tray cool until gel is translucent (takes at least 20 minutes). CLEAN UP ANY DRIPS ON THE BENCH AND RINSE THE BEAKER WITH RO-H2O! |
|
|
|
6. |
Prepare samples as described below. For agarose gels it is advisable to load the same volume into each well. Some samples may need to be diluted with water or TE to achieve this. |
|
|
|
7. |
Turn lever on the caster to break the seal. Carefully remove comb and place casting tray into the electrophoresis box for running. Fill unit with 1X TBE buffer to ~ 1 mm above gel. Pour buffer carefully onto the center of the gel to prevent the gel from sliding off the tray. |
|
|
|
8. |
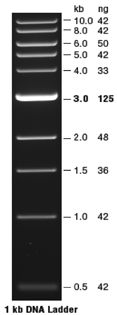
Carefully load 10 µl of the DNA ladder (see below) into a well; load all of each plasmid DNA sample into different wells. Record the order of the samples in your notebook. Do not press the tip into the bottom of the well while loading--allow the sample to sink there. |
|
|
|
9. |
Position the lid and connect the electrodes in the correct orientation. |
|
|
|
10. |
Run gel at 130 V for ~ 30 min. |
|
|
|
11. |
WEAR GLOVES. Place casting tray with gel onto a paper
towel and carefully carry to the photography area. DO NOT
spread EtBr outside the designated area!! From this point
forward, assume that your gloves are contaminated with EtBr.
Do not touch anything that is not supposed
to be contaminated. |
|
|
|
12. |
Photograph the gel and compare the observed bands to the standards (see below). |
|
|
|
13. |
Locate the insert and vector DNA
bands on the agarose gel using long wave (>300 nm) UV
light.
|
|
|
|
14. |
Excise the DNA from the gel using a razor blade or scalpel (use a new one for each piece of DNA; dispose of in a sharps container).
|
|
|
|
15. |
Transfer each gel piece to a sterile 1.5 ml tube; proceed with gel purification of DNA |
|
Copyright, Acknowledgements,
and Intended Use
Created by B. Beason (bbeason@rice.edu), Rice University, 21 November 2007
Updated 22 October 2015